
Quality of Prediction
Leave one out cross-validation for all predictable kinds of biological activity and all substances in PASS training set provides the estimate of PASS prediction accuracy during the training procedure. Average accuracy of prediction is about 95.5% according to the LOO CV estimation, while that for particular kinds of activity varies from 70% (Immunostimulant (HIV)) to 99.9% (VEGFR2 expression inhibitor, Toll-like receptor antagonist, etc.). Estimated accuracy of prediction for all kinds of biological activity predicted by PASS is presented at the web site.
The accuracy of PASS predictions depends on several factors, from which the quality of the training set seems to be the most important one. A perfect training set should include the comprehensive information about biological activities known or possible for each compound. In other words, the whole biological activity spectrum should be thoroughly investigated for each compound included into the PASS training set. Actually, no database exists with information about biologically active compounds tested against each kind of biological activity. Therefore, the information concerning known biological activities for any compound is always incomplete. We investigated the influence of the information's incompleteness on the prediction accuracy for new compounds. About 20,000 "principal compounds" from MDDR database were used to create the heterogeneous training and evaluation sets. At random 20, 40, 60, 80% of information were excluded from the training set. Either structural data or biological activity data were removed in two separate computer experiments. In both cases it was shown that even if up to 60% of information is excluded, the results of prediction are still satisfactory. Thus, despite the incompleteness of information in the training set, the method used in PASS is robust enough to get the reasonable results of prediction.
It must be kept in mind that there exists the fundamental limitation: any observation, estimation, calculation has only restricted accuracy. In absolutely all cases instead of the desirable unknown intrinsic Real value we have only:
Observation = Real + Noise
This is critically important for (virtual) screening especially. To highlight this in Figure 1, the generated data of 1,000 points with binormal distribution and correlation coefficient square R2 = 0,95 and R2 = 0,5 are presented. For R2 = 0,5 it is clear that relationship looks like a weak tendency only. In Figures 2-4 the results of selection of the 100 Bests (with the highest Real values) and the 100 Winners (with the highes Estimation values) among 1,000,000 "screene" examples are presented. It is clear that only for R2 = 0,95 coincidence of the Winners and the Bests is relatively good (about 60%), but for R2 = 0,5 it is practically zero.
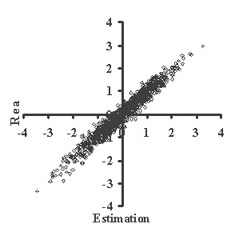 (a)
(a)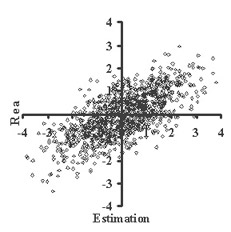 (b)
(b)Figure 1
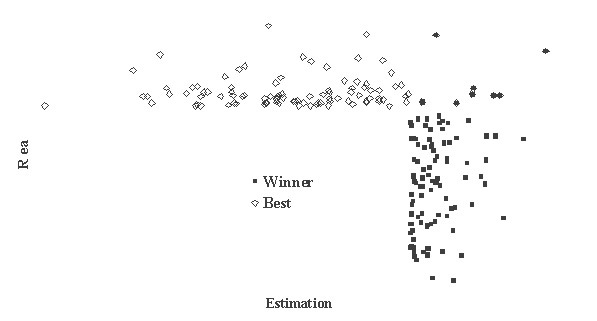
Figure 2
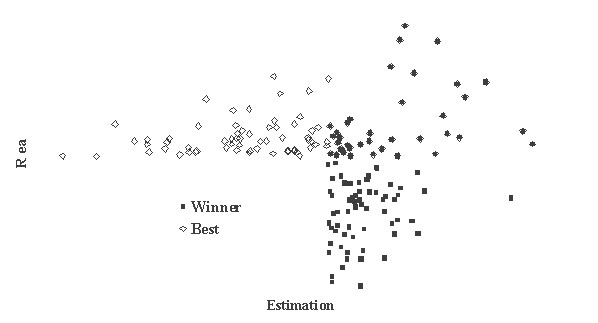
Figure 3
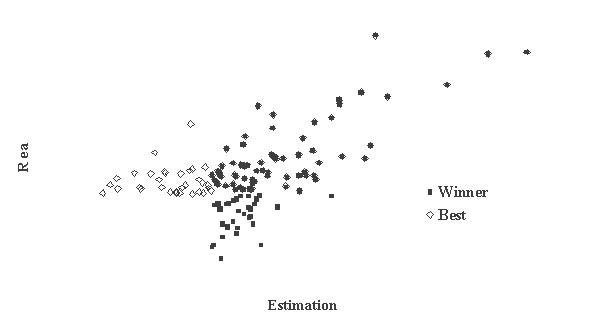
Figure 4
It is possible to perform a complete analysis of such relationships, but even the presented data provide enough evidence for the following conclusion: the method for (virtual) screening must be highly accurate, and/or many different virtual screening methods must be used in combination and/or the number of selected candidates must be sufficiently large at all stages of screening (in Figures 2-4 the number 100 is not "sufficiently large"). Since the predicted with PASS biological activity spectra contain the estimates of probabilities for the main and side pharmacological effects, molecular mechanisms of action and specific toxicity, the choice of the most prospective compounds from the available samples of chemical compounds can be realized on the basis of complex criteria. Both presence of targeted biological effects with desirable mechanisms of action and the absence of unwanted adverse effects & toxicity have to be taken into account. In such studies, the search of leads with the required properties and their optimization to decrease the adverse & toxic effect, usually performed sequentially, will be solved simultaneously. Moreover, it was shown that the algorithms used in PASS can be successfully applied for discrimination between the so-called drug-like and drug-unlike compounds, 1, 3, 4 that provides the possibility for extension of the applicability of the program by "filtering" in early stages chemical compounds, for which probability to become drug is rather small. Evolution of any molecule from hit to lead and from lead to drug-candidate typically is associated with the detailed evaluation of pharmacodynamics and pharmacokinetics of the compound. Using several different probabilistic methods for virtual screening together, it might be possible to increase significantly the rate of promising substances in the selected sub-set. A challenging task is to optimize simultaneously both pharmacodynamics and pharmacokintetics of lead compounds because it is very difficult to modify the appropriate molecular determinants that define the desired compound characteristics, in a consistent manner. However, even this task might be solved using "an integrated software framework that monitors ligand (or library) alterations in the context of 'fitness landscape"'.