About Proteochemometrics Service
Training data preparing
The training set should contain enough reliable data confirmed at the molecular level, requiring careful processing of the information retrieved from available resources. The selected data should present the ligands’ chemical structures, target proteins’ sequences, and the affinity values for each protein–ligand pair. We used the local version of the ChEMBL 28 database, which contains detailed experimentally obtained characteristics of interacting the small molecules and target proteins, including the ligands’ chemical structures, proteins’ peculiarities, and study conditions. To select reliable and suitable data, we formed the MySQL queries with the following limitations:
- Direct testing of the protein–ligand binding;
- The accurate definition of a protein (neither a homolog nor a protein family);
- Testing the isolated protein (it is not in a complex or cellular fraction, etc.);
- Evaluating the interaction by the Inhibitory Constant (Ki) as;
- Excluding the mutant proteins.
Activity Definition
The data retrieved for the same interacting pair often presented the results obtained by different research groups at different experimental conditions. Therefore, we developed a procedure that provided the integration of such data according to certain filtration rules. To evaluate the protein-ligand interaction, we chose Ki, which adopted fixed (e.g. 1.0 µmol) or interval values (>0.1, <50.0 µmol, etc.). We established a cutoff that equals 10.0 µmol and considered the protein and ligand as interacting if the values did not exceed the cutoff. As a result, each protein-ligand pair got the binary interaction index designating the interaction (1) or non-interaction (0).
In the presence of both fixed and interval values, only fixed values were accounted for if even interval values contradicted fixed ones. In the case of the several fixed values, the interaction index adopted a value of 1 if the data median did not exceed the cutoff and 0 otherwise:
- In the absence of fixed values, the procedure does not pass the protein-ligand pair if Ki values were in non-intersected (e.g., <100 and >5000) or limited at two ends (e.g., >100 and <5000)intervals;
- In the case of a few intervals limited below (e.g., >100, >1000, >5000), the index adopted 0 if the maximal edge value was higher than a given cutoff; otherwise, the protein–ligand pair was excluded;
- In the case of a few intervals limited above (e.g., <100, <1000, <5000), the index adopted a value of 1 if the minimal extreme value was less than the cutoff; otherwise, the protein-ligand pair was excluded.
Prediction procedure
Our approach provides extensive opportunities for researchers, maintaining the studies with the three most typical scenarios of in silico assessment the target-ligand interaction.
The 1st scenario presents the common task of structure-activity relationships (SAR) analysis when the model predicts protein targets for a ligand by comparing its chemical structure with structures of compounds with known spectra of targets. Meanwhile, protein names are class-forming features, ignoring any amino acid sequence or 3D structure information.
The 2nd scenario involves the search of small molecule ligands for the test protein by comparing the amino acid sequences with known ligands. Ligands’ identifiers are used as class-forming features without considering their structures.
The 3rd scenario is related to the well-known problem of data incompleteness. In the absence of the information on interaction spectra of both the target and ligand, the predictive algorithm requires the protein sequence and ligand structure to compare them with both training data types.
Prediction of target targets based on the ligand similarity – 1st scenario
Prediction of the ligand’s binding with protein targets uses the protein identifiers as classification features, not considering the sequence similarity. It was done using the PASS software developed to predict the biological activity spectra of organic compounds, successfully applied in cheminformatics and drug design [Filimonov, Poroikov, 2008; Filimonov et al., 2014; Pogodin et al., 2015]. The PASS uses the compounds’ chemical structures in the MOL format, calculating the original descriptors designated as MNA (Multilevel Neighborhoods of Atoms). These descriptors reflect the local structural features responsible for the predicted activities. The PASS predictor is based on the Bayes classifier. It outputs Pa and Pi, values estimating the probabilities of interaction and non-interaction between the target protein and the ligand. So Pa and Pi reflect the structural similarity of test ligands with active and non-active compounds, respectively.
Prediction of protein-ligand interaction according to 2nd and 3rd scenario
Positional scores
The SPrOS method [Karasev et al., 2020a] is applied to predict protein-ligand interactions in the cases of the 2nd and 3rd scenarios. The algorithm uses the positional scores calculated from comparisons between the amino acid sequence of test protein (T) and amino acid sequences of the training set. The fragment similarity score is the sum of similarity scores for matched residue pairs. When matching all F long fragments of the T sequences with all fragments of training sequence k, each position p of T falls into the F fragments and gets the highest fragment scores Spk.
In the current program version, the F value equals 15.
Predictive method
The obtained positional scores are used in the prediction procedure, which also requires the coefficients of ligand specificity [Karasev et al., 2020a; Karasev et al., 2020b;]. The ak value represents a training protein k belonging to the class of proteins interacting with the ligand L.
The bk value represents a protein k belonging to the class of proteins that do not interact with the ligand L.
The following values are calculated to evaluate the interaction between the target T and the ligand L: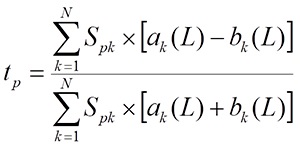
the tp values are averaged on all m positions of the T sequence:
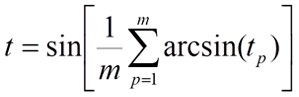
the t0 value is the a priori evaluation of the target-ligand interactions without scoring the sequence similarity:
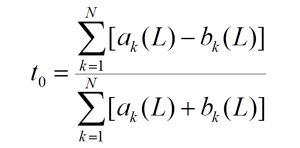
the Binding Estimate (BE) is obtained as follows:
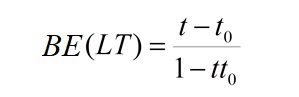
BE(LT) varies from −1 (no interaction) to +1 (interaction).
Prediction according to the 2nd scenario
The binary ak and bk values link the protein targets and ligands within the training set, required to predict the new protein target using training proteins with established ligand spectra. If the training protein k binds the ligand L, ak(L) and bk(L) equal 1 and 0, respectively. If no binding, ak(L) and bk(L) equal 0 and 1. The coefficients for training sets used at this server have been preliminary calculated from the experimentally derived inhibitory constants (Ki) [Karasev et al., 2020a].
Prediction according to the 3rd scenario
The fuzzy belonging coefficients are introduced to predict the new target-ligand interaction under the 3rd scenario if both objects are not characterized [Karasev et al., 2020b]. These coefficients are calculated by the PASS algorithm, likewise 1st scenario. After inputting the test ligand structure, it is compared with all ligands related to each protein from the given training set. The training protein adopts the fuzzy coefficients ak and bk equal to pa and pi values, assessing its interaction with the test ligand before the sequence comparison. The further handling with the protein sequences results in the final estimates of protein-ligand binding. Using fuzzy coefficient allows overcoming the common problem of the training data incompleteness, accounting for hidden positive examples and significantly enriches the training data.
References
Filimonov, D., Poroikov, V. 2008. Chapter 6. Probabilistic approaches in activity prediction, in: Varnek, A., Tropsha, A, (Eds.), Chemoinformatics Approaches to Virtual Screening. RSC Publishing, London, pp. 182 –216
Filimonov, D.A., Lagunin, A.A., Gloriozova, T.A., Rudik, A.V., Druzhilovskii, D.S., Pogodin, P.V., Poroikov, V.V., 2014. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Comp. 50, 444–457.
Karasev, D., Sobolev, B., Lagunin, A., Filimonov, D., Poroikov, V., 2020a. Prediction of protein-ligand interaction based on the positional similarity scores derived from amino acid sequences. Int. J. Mol. Sci. 21, 24.
Karasev, D., Sobolev, B., Lagunin, A., Filimonov, D., Poroikov, V., 2020b. Prediction of protein-ligand interaction based on sequence similarity and ligand structural features. Int. J. Mol. Sci. 21, 8152.
Pogodin, P.V., Lagunin, A.A., Filimonov, D.A., Poroikov, V.V., 2015. PASS Targets: ligand-based multi-target computational system based on a public data and naïve bayes approach. Sar. Qsar. Environ. Res. 26, 783–793.